WASHINGTON (AP) – Die Aufsichtsbehörden der Food and Drug Administration haben am Dienstag ein einzigartiges Medikament für eine seltene Form der Lou-Gehrig-Krankheit zugelassen, obwohl sie weitere Forschung benötigen, um zu bestätigen, dass es den Patienten wirklich hilft.
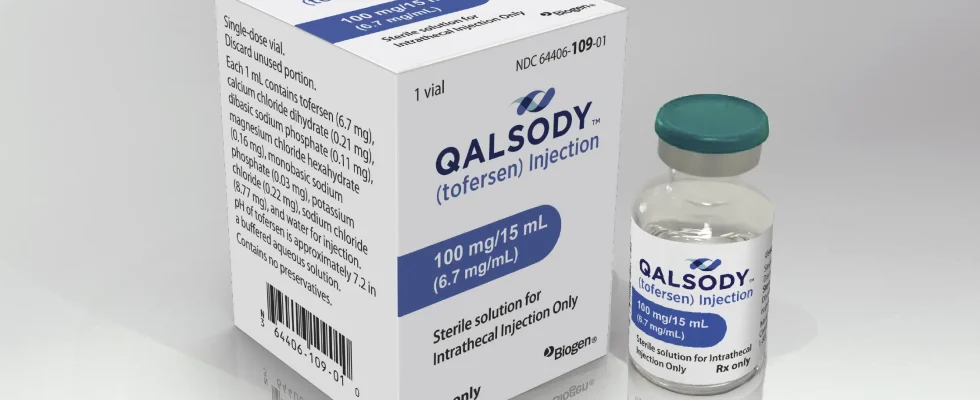
Die FDA hat das injizierbare Medikament von Biogen für Patienten mit einer seltenen genetischen Mutation zugelassen, von der schätzungsweise weniger als 500 Menschen in den USA betroffen sind Funktionen wie Gehen, Sprechen und Schlucken.
Die Zulassung erfolgte über den beschleunigten Weg der FDA, der es ermöglicht, Medikamente auf der Grundlage vielversprechender früher Ergebnisse auf den Markt zu bringen, bevor bestätigt wird, dass sie den Patienten zugute kommen. Diese Abkürzung wird zunehmend von staatlichen Wachhunden unter die Lupe genommen und Ermittler des Kongresses.
Die FDA verlangt von Biogen, das Medikament in einer Studie mit Personen weiter zu untersuchen, die die genetische Mutation tragen, aber noch keine ALS-Symptome haben.
ALS-Patienten hoffen, dass die Entscheidung den Grundstein für beschleunigte Zulassungen zur Bekämpfung der Krankheit legen könnte, von der 16.000 bis 32.000 Menschen in den USA betroffen sind. Die FDA nutzt seit langem die beschleunigte Zulassung, um die Verfügbarkeit von Medikamenten gegen Krebs und andere tödliche Erkrankungen zu beschleunigen.
Das Medikament Tofersen soll die genetischen Botenstoffe blockieren, die eine toxische Proteinform produzieren, von der angenommen wird, dass sie die Krankheit bei etwa 2 % der ALS-Patienten antreibt. Biogen mit Sitz in Cambridge, Massachusetts, wird es unter dem Markennamen Qalsody verkaufen. Die Patienten erhalten drei initiale spinale Injektionen des Medikaments über einen Zeitraum von zwei Wochen, gefolgt von einer monatlichen Dosis. Die häufigsten Nebenwirkungen im Zusammenhang mit dem Medikament waren Schmerzen, Müdigkeit und erhöhte Rückenmarksflüssigkeit.
Die 100-Personen-Studie von Biogen konnte nicht zeigen, dass das Medikament die Krankheit im Vergleich zu einer Scheinbehandlung signifikant verlangsamte. Die Patienten wurden mehr als sechs Monate lang mit einer Skala verfolgt, die den Rückgang grundlegender Bewegungen misst, einschließlich Schreiben, Gehen und Treppensteigen.
Aber diejenigen, die Tofersen erhielten, zeigten signifikante Veränderungen in den Spiegeln des toxischen Proteins und einer zweiten neurologischen Chemikalie, die als Schlüsselindikator für das Fortschreiten der Krankheit gilt.
„Die Ergebnisse lassen mit ziemlicher Wahrscheinlichkeit einen klinischen Nutzen für Patienten vorhersagen“, sagte die FDA in einer Erklärung zur Bekanntgabe der Zulassung.
Letzten Monat stimmte ein externes Gremium von FDA-Beratern einstimmig dafür, dass diese Änderungen eine bedingte Zulassung rechtfertigen, während weitere Daten gesammelt werden, um den Nutzen des Medikaments zu bestätigen. Dasselbe Gremium sagte, dass die aktuellen Daten von Biogen, einschließlich der fehlgeschlagenen Patientenstudie, nicht stark genug seien, um eine vollständige Genehmigung zu rechtfertigen.
Die FDA-Regulierungsbehörden sind befugt, eine beschleunigte Zulassung zu erwirken von Medikamenten, die ihr erwartetes Versprechen nicht erfüllen, wenn auch bis vor kurzem, sie nutzten diese Macht selten. In den letzten Jahren hat die FDA ihre Bemühungen verstärkt, unbewiesene Medikamente vom Markt zu verdrängen, angesichts der Kritik, dass zu viele teure, unwirksame Medikamente jahrelang verfügbar sind.
Gleichzeitig hat die FDA bei der Zulassung von Arzneimitteln für seltene und schwächende neurologische Erkrankungen, einschließlich Alzheimer, eine erhöhte „regulatorische Flexibilität“ gezeigt und ALS.
Im September erteilte die FDA die volle Zulassung zu einem anderen ALS-Medikament, basierend auf einer kleinen Studie im mittleren Stadium, in der die Patienten scheinbar langsamer Fortschritte machten und mehrere Monate länger überlebten. Normalerweise verlangt die FDA zwei große Studien oder eine Studie, die auf eine „sehr überzeugende“ Verbesserung der Überlebensrate hindeutet.
Einige Versicherer haben nur begrenzten Zugang zu dem neuen MedikamentRelyvrio, unter Berufung auf den ungewissen Nutzen und die Kosten von 158.000 USD pro Jahr.
Biogen gab am Dienstag keinen Preis für sein Medikament bekannt, sagte jedoch, es werde „mit anderen kürzlich eingeführten ALS-Behandlungen vergleichbar sein“.
Die ALS Association und andere Patientengruppen begrüßten die Zulassung.
„Dies ist das zweite Mal in weniger als einem Jahr, dass unsere Gemeinschaft die Zulassung eines neuen Medikaments zur Behandlung von ALS feiern kann, und wir haben große Hoffnung für die Zukunft“, sagte Calaneet Balas, Präsident und CEO der Gruppe.
Seit der letztjährigen Zulassung von Relyvrio haben ALS-Patienten und Befürworter weiterhin Druck auf die FDA ausgeübt weitere Behandlungen in Betracht ziehen für die Krankheit. Dazu gehört eine experimentelle Stammzellenbehandlung des winzigen Arzneimittelherstellers Brainstorm Cell Therapeutics.
In einem seltenen Schritt hat die FDA kürzlich zugestimmt, ein öffentliches Treffen über die Behandlung abzuhalten, obwohl sie sich zuvor geweigert hatte, den Antrag des Unternehmens zu prüfen, unter Berufung auf fehlgeschlagene Ergebnisse seiner Hauptstudie.
Die FDA hat jetzt vier Medikamente für ALS zugelassen, von denen nur eines nachweislich das Leben verlängert. Die Krankheit zerstört nach und nach Nervenverbindungen, die für grundlegende Bewegungen und – schließlich – für die Atmung benötigt werden. Es gibt keine Heilung und die meisten Menschen sterben innerhalb von drei bis fünf Jahren nach der Diagnose.
___
Folgen Sie Matthew Perrone auf Twitter: @AP_FDAwriter
___
Das Associated Press Health and Science Department erhält Unterstützung von der Science and Educational Media Group des Howard Hughes Medical Institute. Für alle Inhalte ist allein der AP verantwortlich.